

















Historically, injectable drugs havemostly been packaged aseptically, with pharmaceutical laboratoriescarrying out the sterilization of the primary packaging components internally.
The conventional solution is for pharmaceutical laboratories to sterilize components before use, generally using steam.Yet, the alternative solution is to use packaging components which are sterilized beforehand, by manufacturers. In this instance, the syringe is sterilized with ethylene oxide after assembly of the tip cap’s needle shield. Thesyringe’srubber plunger is thensterilized by gamma irradiation, which has proved to be an efficient means of sterilizing packaging components.
This case study by Aptar Pharma explores how gamma sterilization effectively provides pharmaceutical laboratories with all the necessary safety requirements, to ensure product release.
Ready-to-Use gamma sterilized components
The transfer of washing and sterilization operations from the laboratory to the plunger supplierhas several advantages for pharmaceutical laboratories:
- Reduction in humanreliance
- Improvedproductivity
- Economic advantages – reduced need to invest in equipment
- Reduced stock levels
Addressing issues of risk
Ionizing radiation has the advantage of sterilizing syringe plungers within their packaging.The risk of contamination is reduced to the handling stage, when components are transferred to the sterile zone of drug product fill & finish. At this stage, the exterior of the double packaging is chemically decontaminated before removal. Equally, containers equipped with double-port transfer systems (Rapid Transfer Port – RTPs) can be used for the aseptic transfer of materials and components in isolators and RABS.
Gamma rays are highly penetrating and can be used for treating whole pallets, with the added benefit that exposure dose is well controlled and can be easily recorded.
Ensuring a regulatory environment
The Pharmaceutical cGMPs Guidance for Industry states that containers and closures should be rendered sterile and, for parenteral drug products, nonpyrogenic. The process used depends on the container type or its closure material, and written evidence must specify the validation and revalidation of these processes.
The sterilization of rubber plungers requires the close involvement of three different parties: the elastomeric component manufacturer, the sub-contractor performing the sterilization and the pharmaceutical laboratory using the sterilized components.
Compatibility of the elastomer formulation
Gamma irradiation can lead to the degradation of some materials. Its influence on the rubber formulation properties must be studied to ensure the performance characteristics of the plungersare not affected by the sterilization.
The irradiation influence on a given property can be different depending on the formulation. Yet, the resistance to gamma sterilization can be modified by the selection of a suitably-designed rubber formulation. Some formulations are more sensitive to radio sterilization, and the evolution of the property affected is not necessarily proportional to the level of radiation received. Predicting the influence of ionizing radiation on a given type of rubber is therefore impossible without research.
The influence of gamma radiation
The influence of gamma irradiation must be assessed with regards to Figure 1:
- The mechanical, chemical and functional properties of the formulation
- The biological properties of the formulation for irradiation levels of 25 and 50 kGy, corresponding to one and two radiosterilizationsrespectively, according to the recommendations of the standard pharmacopoeias.
Figure: 1
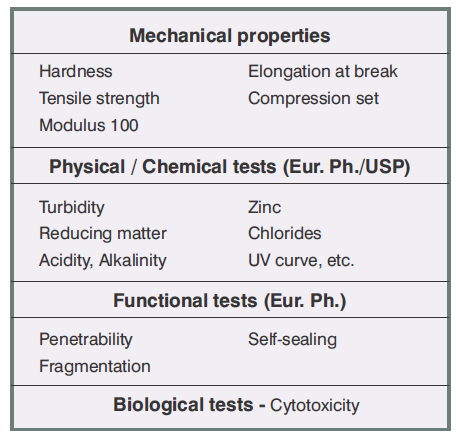
Sterile plungers: Validation
Obtaining “Ready-to-Use” syringe plungers requires control of all applicable pharmaceutical requirements, which mainly affect the finishing steps (washing and sterilization). The manufacturing process must be validated to ensure the required quality.
Validation of washing procedures
Historically, preparation of pharmaceutical plungers has been carried out by pharmaceutical laboratories themselves. These procedures for preparing primary packaging components are precisely described in the registration files of each injectable drug which has been registered with both the FDA and the European regulatory authorities. These operations are now largely undertaken by component manufacturers who market “Ready-to-Sterilize” products.
Regulatory requirements concern mainly the quality of the fluids used, the environment and thevalidation of the procedure. The quality of water to be used is clearly defined in the Guidelines issued by the FDA and the EMEA12. In both cases, it must comply with the “Purified Water” or “highly purified water” Monographs for the washing and first rinsing operations, and the “Water for Injection” Monograph for the final rinsing.
The environment should not allow any re-contamination after washing. The sensitive item being packaged should take place in an ISO 5 standard classified zone. To meet these requirements, Aptar Pharma uses the UltraClean 6 evolutionwashing process for plungers,obtaining the highest particulate and microbiological cleanliness.
Validation is divided into three successive phases.
- Installation Qualification (IQ) – Checks all the equipment is installed according to the manufacturer’s specifications
- Operational Qualification (OQ) – Demonstrates that equipment functions as anticipated under normal and extreme usageconditions.
- Performance Qualification (PQ) – Shows that the process allows achievement of predetermined product specification.
Testing particulate and microbiological cleanliness does not just rely on the washing procedure. All manufacturing processes related to plungers must be optimized to fully limit contamination risks. This optimization allows contamination below 0.1 CFU/cm2 before sterilization to be absolutely guaranteed.
Aptar Pharma has prepared a unique DMF for its production sites, ensuring the same quality is provided at every source.
Validation of sterilization procedures
Validation of the sterilization procedures with gamma irradiation has four steps:
Step 1:Determining the maximum dose
Determining the permitted maximum irradiation dose concerns both the product itself and its packaging. The most relevant tests are described in the European Pharmacopoeia, applicable to rubber closures3. They reveal the effect of gamma sterilization on the product’s chemical and functional properties. These tests may be augmented by measuring the mechanical properties in standardized test samples.
The results obtained from chlorobutyl rubber closures (Figure 2) show great stability of all properties studied after exposure to 25 and 50 kGy. Similar results are obtained with bromobutyl rubber formulation tailored for irradiation.
The elastomer-based formulations offered by AptarPharma for sterile plungers are designed to be compatible with radiosterilization. Exposing the plungers to 25 and 50 kGy either does not affect the investigated properties or has a negligible influence on these properties. Even when irradiated and aged, these formulations do not approach the limits described in the principal norms and pharmacopoeias.
The influence of ageing was also studied. The tests listed in Figure 1 were performed after one and two years. No notable change in the chemical profile of the elastomer formulation was observed.
Figure: 2
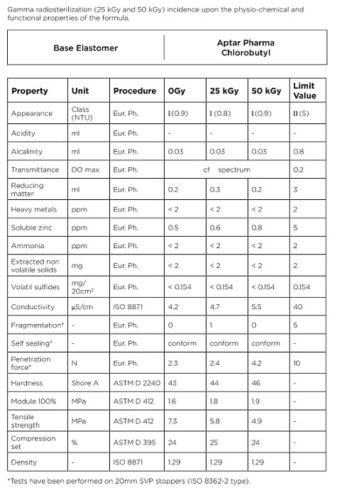
Step 2: Determination of the sterilizing dose
The following stage involves selecting the sterilizing dose. To perform this task, Aptar Pharmahas chosen to use ISO regulation 111374 relating to the sterilization of medical devices and, more specifically, to use Method 1, which involves working from information about bioburdens.
A determination of the bioburden is made from three different batches of syringe plungers and a table provides the dose at which SAL (Sterility Assurance Level) is 10-2for this bioburden. This value is then used as the verification dose. A sample of 100 syringe plungers is then exposed to this verification dose and the sterility of each product is tested individually. If there are less than two positive tests out of 100, the sterilizing dose where SAL is 10-6is determined using the same table.
Once the dose is determined, a periodic audit must be conducted to confirm the validity of this sterilizing dose.

Step 3: Determining dose mapping
Gamma sterilization permits full pallets of product to be processed.
Therefore, the first step consists ofdetermining the configuration which allows for the most homogenous irradiation dose possible to be obtained between the different points within the load being treated. Dose-mapping is validated from three different loads.
Validation involves distributing the dosimeters to different points within each load and therefore determining the position of the “cold point (minimum dose)”and the position of the “hot point (maximum dose)” so that exposure relating to these points can be compared to a ‘routine’ or reference point. Afterwards, a single dosimeter will be necessary for each load and will be placed at the ‘routine point’.
From the irradiation dose measured at the routine point we can deduce the doses received at both the“cold”and “hot”points and, therefore, ensure that each point within the load has at least received the sterilizing dose without exceeding the set maximum dose.
Step 4: Determining the expiration date
The expiration date is determined following an aging study conducted on the finished product. This allows packaging integrity to be checked alongside the behavior of the irradiated product. Packaging integrity is essentialin terms of preserving sterility.The packaging of the sterilized components must provide protection throughout its shelf life. This may be demonstrated by checking the vacuum level in the bags prior to use.
Conclusion
Supplying “Ready-to-Use” primary packaging materials is the next logical step resulting from the supply of“Ready-to-Sterilize” components. Gamma sterilizationprovides the pharmaceutical laboratories with all the necessary safety requirements. The issue of responsibility can be settled within a contractual context where the key points to consider are the validation procedure, the validation files and the data leading to the product being released.
References
(1) FDA Guidance for Industry. Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice. September 2004.
(2) EMEA Note for Guidance on Quality of Water for Pharmaceutical Use. May 2002.
(3) European Pharmacopoeia 5.0. 3.2.9. Rubber Closures for Containers for Aqueous Parenteral Preparations, for Powders and for Freeze-Dried Powders.
(4) ISO 11137-2:2013 Sterilization of health care products — Radiation — Part 2: Establishing the sterilization dose





