

















Microheterogeneity in seemingly homogenous cell populations plays a key role in determining the pathways pursued by biological systems. Variation in expression of gene products and other processes result in this underlying microheterogeneity. Understanding these variations at the single cell level should help in identifying the few cells that act as a seed for disease, such as in cancer development.
Studying DNA and RNA molecules within the cell is one of the most common approaches to study single cell biology and has also helped motivate the measurement of proteins at the single cell level. Exponential advances have been achieved in single cell DNA and RNA sequencing technologies and depending on the application, a variety of sequencing techniques can be employed for these studies.[1]
With the help of such advanced technologies, studies involving the measurement of the single cell transcriptomes of more than a million individual cells are now feasible[2] and have revealed previously unseen biology and highlighted the heterogeneity of single cells, thereby opening up new areas of biology and medicine. A common enabling factor in all these approaches is the ability to amplify DNA and RNA molecules to virtually any desired amount, bringing it into a detectable or quantifiable range.[3]
Protein function
Genetic information contained in the genome is translated to RNA which is used as a template for the production of proteins which in their turn shape the metabolome. However, the proteome contains additional layers of complexity that are not obviously hardcoded in the genome. Protein function is frequently modulated through post-translational modifications (PTMs) such as phosphorylation and ubiquitylation. This can change the functional course of the cell with very fast kinetics (Figure 1), and processes such as endogenous proteolysis and glycosylation are known to play a key role in normal biological function as well as in abnormal process such as oncological mechanisms.[4],[5]
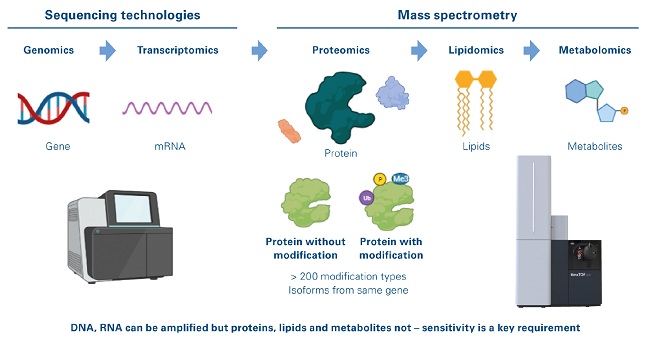
Figure 1: Single cell-omics landscape
The direct detection of proteins, lipids, and metabolites from single cells in an unbiased manner has seemed out of reach until recently. Unbiased proteomics of single cells have been performed in the recent years by specialized research groups involving nano-fluidics that focused on minimizing the loss during sample preparation and multiplexing samples to boost signal intensity.[6],[7] However, despite these solutions, the field still has needed innovations that can boost the sensitivity of the mass spectrometer.
Technological advancements
Epiproteomics is the study of post-translational modification of proteins, which includes more modification of proteins by more than 200 biochemical moieties, the presence of which can be measured in an unbiased fashion only using mass spectrometry (MS). Proteins, lipids and metabolites cannot be amplified and require highly sophisticated and sensitive mass spectrometers for their direct detection at the single cell level.
One technological advancement is trapped ion mobility spectrometry (TIMS), a separation technique in gas phases where ions are propelled through the TIMS tunnel by a gas flow. An electrical field controls each ion from moving beyond a position defined by the ion’s mobility, where the push it experiences from the gas flow matches the force of the electrical field. Ramping down the electrical field allows selectively releasing ions from the TIMS tunnel according to their mobility. TIMS allows researchers to reproducibly measure the collisional cross-section (CCS) values for all detected ions. CCS is a fourth measurement dimension added to the mass, LC retention time and MS/MS spectra usually measured in proteomics experiments resulting in what is now termed 4D-Proteomics. CCS values and the TIMS separation can be used to further increase the system’s selectivity, enabling more and more reliable relative quantitation information from complex samples using short gradient analyses.
The introduction of TIMS enabled the development of the novel parallel accumulation serial fragmentation (PASEF)[8] acquisition method allows for ions to be accumulated in the front section, while ions in the rear section are sequentially released depending on their ion mobility for very high-speed MS and MS/MS analysis. The combination of TIMS and PASEF provided a spectroscopic technique used with liquid chromatography coupled to mass spectrometry (LC-MS) based proteomics to improve sequencing speed and sensitivity. PASEF makes efficient usage of the ion beam and, together with intelligent precursor selection of ions eluting from a TIMS cycle, achieves rapid MS/MS identification speed. In addition, the ions are focused in space and time within the TIMS cell, resulting in a significant boost in the sensitivity. This enables the analysis of low sample amounts in the range of low nanogram peptide loads.
TIMS measurements also provide CCS values and separation of isomeric species that are mobility offset but mass aligned and alleviates ratio compression in multiplexed quantification approaches. By using a dual TIMS analyzer, ions may be nearly continuously queued for accumulation, sorting and elution by mobility, allowing a duty cycle near 100% (Figure 2). Achieving this depth offers completely new possibilities to analyze large sample cohorts of hundreds to thousands of samples.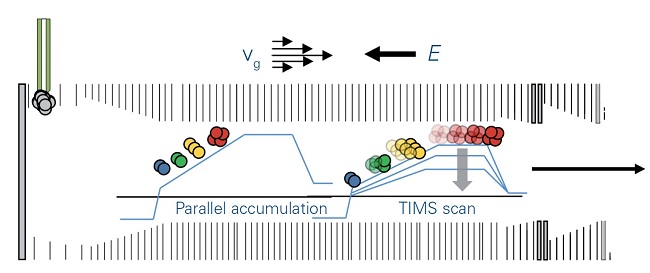
Figure 2: TIMS operation mode
With the directed forces of gas flow, vg, and electrical field, E, ions are accumulated and trapped according to mobility. Stepwise decrease of the electrical field permits serial elution of separated ions, which are then transferred to the QTOF mass spectrometer. Using the parallel accumulation operation mode, ions are eluted from the second part of the TIMS device while the next series of ions is introduced into the first part of the TIMS device.
TIMS instrumentation
The introduction of the timsTOF Pro platform (Bruker, Billerica, Massachusetts) resulted in two paradigm shifts in the proteomics community. The amount of peptide required for standard proteomics analysis was reduced by an order of magnitude (Figure 3) and the time required for standard proteomic analysis was reduced by a factor of four. In a recent study, qualitative and quantitative information for up to 1400 proteins was obtained from single cells using an “Unbiased Single Proteomics” approach which did not require complex isobaric labelling chemistries to boost the peptide signals.[9] Cluster analysis of the data could distinguish cell types and cell cycle stages despite the technology not specifically targeting known and verified markers. With the latest generation, the timsTOF Pro 2, it is now possible to detect ~8000 proteins in little more than an hour of analysis time from only 200 ng of starting material without sacrificing any sequence coverage of these proteins.[10]
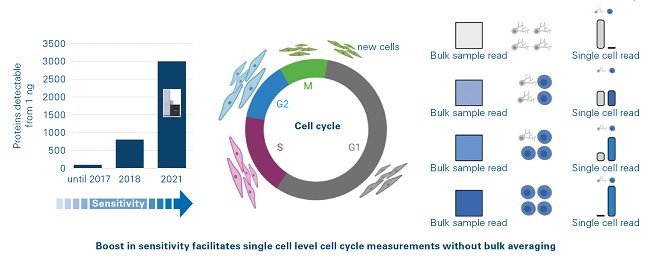
Figure 3: Ultimate sensitivity of timsTOF platform
The timsTOF SCP can be programmed to acquire data in advanced modes such as standard data-dependent acquisition (DDA) PASEF or data-independent acquisition (DIA) PASEF, which allow for the unbiased detection and label free quantification of proteomes. Alternatively, the instrument can also be run in a targeted acquisition mode called parallel reaction monitoring (PRM) PASEF, which can target specific panels of proteins analogous to mass cytometry technology but without the requirement for affinity reagents. Thus, the timsTOF SCP can discover new markers of cellular behavior as well as complementing existing technology for targeted detection.
Conclusion
Single cell proteomics technologies are now entering the mainstream, thanks to the pioneering work of a relatively small group of dedicated scientists and the emergence of extremely sensitive mass spectrometers. These technologies have the potential to transform our understanding of cell biology at the macromolecular level and answer fundamental questions regarding protein dynamics, cell differentiation trajectories, and mechanisms of disease. While these processes act at the nano- and microscopic level, they also fundamentally influence higher order macroscopic behavior. Hence it is vital that these processes are understood at the highest possible spatial resolution.
The introduction of 4D-proteomics capabilities has bridged the gap between the requirements of the most demanding proteomics approaches, such as clinical research proteomics, companion diagnostics research and personalized medicine research – and the solutions effectively available on the market.
References:
[1] Chen et al. Front. Genet., 05 April 2019 | https://doi.org/10.3389/fgene.2019.00317.
[2] Cao, J., Spielmann, M., Qiu, X. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). https://doi.org/10.1038/s41586-019-0969-x.
[3] de Bourcy CFA, et al. (2014) PLoS ONE 9(8): e105585. https://doi.org/10.1371/journal.pone.0105585.
[4] Villanueva et al. J Clin Invest. 2006;116(1):26–30. https://doi.org/10.1172/JCI27467.
[5] Connelly, M.A., Otvos, J.D., Shalaurova, I. et al. J Transl Med 15, 219 (2017). https://doi.org/10.1186/s12967-017-1321-6.
[6] Hartlmay, D. et al. bioRxiv 2021.04.14.439828; doi: https://doi.org/10.1101/2021.04.14.439828.
[7] Slavov, N. Current Opinion in Chemical Biology [Internet]. 2021;60 :1 – 9.
[8] Meier F, et al. (2018), Molecular & Cellular Proteomics. 17(12), Dec. 01, 2018.
[9] Brunner, A-D et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. bioRxiv 2020.12.22.423933; doi: https://doi.org/10.1101/2020.12.22.423933.
[10] https://www.bruker.com/en/products-and-solutions/mass-spectrometry/timstof/_jcr_content/root/contentpar/search_copy.download-asset.pdf/776a1cc2-356e-4d45-a5c6-13b44f897678/1888579-lcms-184-fast-proteomics-ebook.pdf



